STUDY OVERVIEW
WHY TYVASO?
SO
your patients can benefit from the groundbreaking research conducted by United Therapeutics1,2

The INCREASE trial demonstrated the efficacy and safety of TYVASO (inhaled treprostinil) for patients with PH-ILD, making treatment possible for the first time1-3*
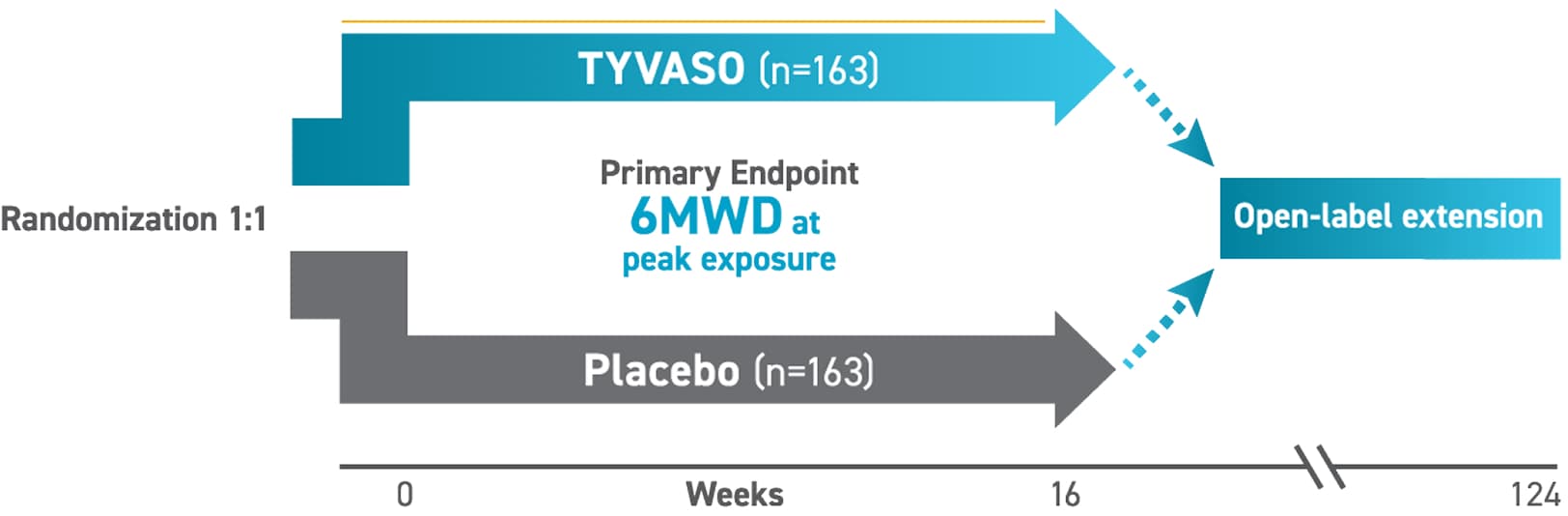
*In the INCREASE trial, the starting dose was 3 breaths, 4x daily (during waking hours) with the TYVASO nebulizer. Dose escalations (1 breath, 4x daily) could occur up to every 3 days, with a target maintenance dose of 9 breaths, 4x daily, and a maximum of 12 breaths, as clinically tolerated.2,3
TYVASO was evaluated across multiple clinical endpoints
Primary Endpoint2
- Change in 6MWD measured at peak exposure from baseline to week 16 (between 10 and 60 minutes after most recent dose)
Secondary Endpoints3,5
- Change in NT-proBNP from baseline to week 16
- Time to first clinical worsening event—from time of randomization until study discontinuation (1 of the following criteria met):
- Hospitalization due to a cardiopulmonary indication
- Decrease in 6MWD >15% from baseline directly related to disease under study at 2 consecutive visits and ≥24 hours apart
- Death (all causes)
- Lung transplantation
- Change in peak 6MWD at week 12
- Change in trough 6MWD at week 15
- ≥4 hours after the most recent study drug dose and ≥24 hours prior to week 16 6MWD
Select Safety Endpoints4†
- AEs
- Oxygenation:
- Pulse oximetry (SpO2)
- Supplemental oxygen requirement (L/min)
- PFTs‡:
- Forced vital capacity (FVC)
- Forced expiratory volume in 1 second (FEV1)
- Total lung capacity (TLC)
- Lung diffusion capacity (DLCO)
- Exacerbations of underlying lung disease§
Based on a concern that treating WHO Group 3 pulmonary hypertension with pulmonary vasodilators could worsen V/Q matching in patients, PFTs (including FVC) and exacerbations were included as safety endpoints due to the potential risk of V/Q mismatch.5
Key Inclusion Criteria4
Patients aged ≥18 were eligible to enroll if they had:
- A confirmed diagnosis of WHO Group 3 PH based on CT imaging within 6 months prior to randomization and demonstrated evidence of any form of ILD or CPFE
- Right heart catheterization within 1 year prior to randomization, with the following documented parameters:
- Pulmonary vascular resistance (PVR) >3 WU
- Pulmonary capillary wedge pressure (PCWP) of ≤15 mm Hg
- Mean pulmonary arterial pressure (mPAP) of ≥25 mm Hg
- Baseline 6MWD ≥100 m
- A stable and optimized dose for ≥30 days prior to randomization if on a chronic medication for underlying lung disease (ie, pirfenidone, nintedanib)
- Baseline FVC of <70% in patients with connective tissue disease
Key Exclusion Criteria4
Patients were not eligible to enroll if they had:
- Diagnosis of PAH or PH for reasons other than WHO Group 3 PH-ILD
- Received any PAH-approved therapy within 60 days of randomization
- Evidence of clinically significant left-sided heart disease (PCWP >15 mm Hg and LVEF <40%)
- Received >10 L/min oxygen supplementation by any mode of delivery at baseline
- Exacerbation of underlying lung disease or active pulmonary or upper respiratory infection within 30 days of randomization
- Initiation of pulmonary rehabilitation within 12 weeks prior to randomization
- Acute pulmonary embolism within 90 days of randomization
Baseline characteristics of the INCREASE study population (N=326)2-4

| Time since PH-ILD diagnosis | 6.5 months (mean) |
|---|---|
| Supplemental oxygen | 71.5% |
|
Chronic medication for underlying lung disease|| |
|
| 6MWD | 259.6 m (mean) |
| NT-proBNP | 503.9 pg/mL (median) |
| FVC (% predicted) | TYVASO group: 62.5% (mean) Placebo group: 63.8% (mean) |
| DLCO (% predicted) | TYVASO group: 30.0% (mean) Placebo group: 28.1% (mean) |
‡Additional safety endpoints included clinical laboratory parameters, vital signs, electrocardiograms, and hospitalizations due to a cardiopulmonary indication.4
§PFTs were prespecified as safety assessments; however, based on study results, exploratory post hoc efficacy analyses were conducted.1,3
||An acute, clinically significant respiratory deterioration characterized by evidence of new widespread alveolar abnormality determined by the investigator.3
¶At baseline, 54% of the patients with IPF were taking pirfenidone or nintedanib. When the INCREASE trial was enrolling patients, both pirfenidone and nintedanib were only indicated for use in IPF.4,6-8
6MWD=6-minute walk distance; AE=adverse event; CHP=chronic hypersensitivity pneumonitis; CPFE=combined pulmonary fibrosis and emphysema; CT=computed tomography; CTD=connective tissue disease; DLCO=diffusing capacity of the lung for carbon monoxide; FVC=forced vital capacity; IIP=idiopathic interstitial pneumonia; ILD=interstitial lung disease; IPF=idiopathic pulmonary fibrosis; LVEF=left ventricular ejection fraction; NT-proBNP=N-terminal pro−B-type natriuretic peptide; PAH=pulmonary arterial hypertension; PCWP=pulmonary capillary wedge pressure; PFT=pulmonary function test; PH=pulmonary hypertension; RCT=randomized controlled trial; SpO2=saturation of peripheral capillary oxygenation; V/Q=ventilation/perfusion; WHO=World Health Organization; WU=Wood units.
The information contained in this section of the site is clinical in nature and specifically created for healthcare professionals. If you are not a US healthcare professional, please click CLOSE to return to the consumer section of the site.